MHC-I binding predictions - Tutorial
Guidelines for selecting thresholds (cut-offs) for MHC class I and II binding predictions can be found
here.
How to obtain predictions
This website provides access to predictions of peptide binding to MHC class
I molecules. The screenshot below illustrates the steps necessary to make a prediction.
Each of the steps is described in more detail below.
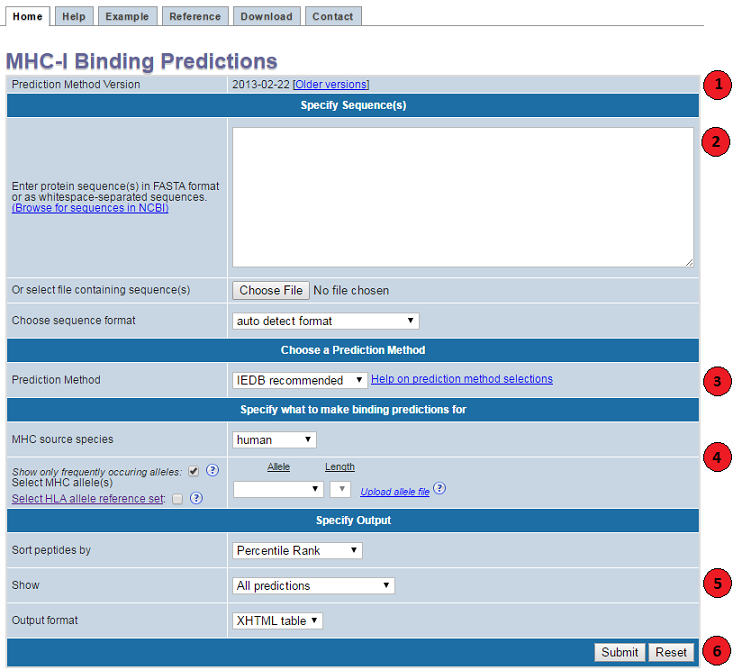
1. Specify prediction method version
The provided date indicates when the tools were released.
2. Specify sequences
First specify the sequences you want to scan for binding peptides. The sequences
should either be entered directly into the text area field labeled "Enter protein sequence(s),
or can be taken from a file that has to be uploaded using the button labeled "Browse". Please
enter no more then 200 FASTA sequences or upload file size less than or equal to 10 MB per query.
The sequences can be supplied in three different formats:
- Space separated sequences
- One continuous sequence
- FASTA format
The format of the sequences can be specified explicitly using the list box
labeled "Choose sequence format". If that list box is set to "auto detect format", the
input will be interpreted as FASTA if an opening ">" character is found, or as
a continuous sequence otherwise.
All sequences have to be amino acids specified in single letter code (
ACDEFGHIKLMNPQRSTVWY)
3. Choose a prediction method
The prediction method list box allows choosing from a number of MHC class I binding prediction methods:
Artificial neural network (ANN),
Stabilized matrix method (SMM),
SMM with a Peptide:MHC Binding Energy Covariance matrix (SMMPMBEC),
Scoring Matrices derived from Combinatorial Peptide Libraries (Comblib_Sidney2008),
Consensus,
NetMHCpan,
NetMHCcons,
PickPocket and
NetMHCstabpan.
IEDB recommended is the default prediction method selection and is updated periodically based on the availability of predictors and observe predicted performance for a given allele. Currently for peptide: MHC-I binding prediction, NetMHCpan EL 4.1 is used across all alleles. For prior versions of the 'IEDB recommended' method (2.22 and earlier), the Consensus method is used if available for the molecule, otherwise NetMHCpan is used.
NetMHCpan versions 4.0 and above include separate predictors for binding (BA) and elution (EL). While BA predictions evaluate the ability of a peptide to bind an MHC molecule, EL predictions also incorporate the likelihood that the peptide will be naturally processed and presented, which makes it more likely to be recognized as a T cell epitope. As these methods have distinct purposes and performance characteristics, as of September 2023, the IEDB has divided the recommended methods according to the intended purpose of the prediction. The
weekly automated benchmarks for binding predictions consistently show that NetMHCpan 4.1 BA is the top-performing binding predictor. It is anticipated that the forthcoming automated benchmarks for methods trained on elution data will indicate that NetMHCPan 4.1 EL here is the top performer.
Recommended Methods used in the tool:
| IEDB Tools Version | recommended method |
|---|
| 2023.09 (current) | NetMHCPan 4.1 EL (epitope prediction) |
| NetMHCPan 4.1 BA (binding prediction) |
| 2020.09 | NetMHCPan 4.1 EL |
| 2020.04 | NetMHCPan 4.0 EL |
| 2.22 and earlier | Consensus, if available; otherwise, NetMHCpan |
Automated benchmarks: Of note, we fully expect the IEDB recommendation to change as we perform larger benchmarks
of newly developed methods on blind datasets to determine an accurate assessment of prediction quality.
Towards that end, automated benchmarks have been established to continuously evaluate the performance of the existing
MHC class I binding methods. These benchmarks are updated weekly as new datasets are deposited into
the
IEDB and the latest results can be found
here.
Method versions used in the tool:
| Method | Version | Source |
|---|
| NetMHCpan EL | 4.1 | DTU |
| NetMHCpan EL | 4.0 | DTU |
| NetMHCpan BA | 4.1 | DTU |
| NetMHCpan BA | 4.0 | DTU |
| NetMHC (ANN) | 4.0 | DTU |
| NetMHCcons | 1.1 | DTU |
| PickPocket | 1.1 | DTU |
| NetMHCstabpan | 1.0 | DTU |
| SMM | 1.0 | LJI |
| SMMPMBEC | 1.0 | LJI |
| PickPocket | 1.1 | LJI |
| Comblib_Sidney2008 | 1.0 | LJI |
4. Specify what to make predictions for
Predictions are not limited to peptides of one specific length binding to one specific allele, but
multiple allele/length pairs can be submitted at a time.
The allele / peptide length combination can be selected using the
list boxes in this section.
For some allele / peptide length combinations, no prediction tools exist
because there is too little experimental data available to generate them.
For instance, selecting an MHC source species of human will allow you to
select a distinct set of MHC alleles and Peptide lengths related to the human
MHC source species. Alternately, selecting a MHC source species of mouse
will allow you to select a different set of MHC alleles and Peptide lengths
related to the mouse MHC source species.
Selections in the listboxes in this section influence the values available in others.
For example, selecting "mouse" as the MHC source species will limit
the selections available in the MHC allele listbox. Similarly, the allele chosen will
limit the available peptide lengths.
• Frequently occurring alleles:
By default "Show only frequently occurring alleles" check-box is checked. This allows the selection
of only those alleles that occur in at least 1% of the human population or allele frequency of 1% or higher.
However, un-checking the check-box will allow selection of all the alleles and corresponding peptide lengths
for a particular species.
• Format for the upload allele file:
File should be in simple text format containing comma separated values, where each allele is separated from it's length by a comma followed by a new line.
Therefore, you can upload a file with each line containing only one such allele-length pair(example given below). However, you may also
choose allele(s) and their length(s) from the drop-down selection in together with your uploaded file.
Example:
HLA-A*02:01,9
HLA-B*15:01,9
HLA-A*02:06,10
...
Additional information regarding HLA allele
frequencies and
nomenclature are also provided.
Note: for NetMHCpan method, there is an option to paste a single full length MHC protein sequence in FASTA format,
instead of selecting alleles from the dropdown list.
• Select HLA allele reference set:
When the IEDB recommended option is selected, this box can be checked to select a reference panel of 27 alleles, as described
here.
5. Specify the output
The menus in this section change how the prediction output is displayed.
To limit the number of results displayed, which can significantly speed up the
time it takes to make a prediction, it is possible to define an upper boundary for
the prediction in the "cutoff" field. Note that the listbox preceding the "cutoff" field
has to be set to "Predicted IC50 [nM] " for the cutoff to take effect.
To reuse the prediction results in an external program, it is possible to
retrieve the predictions in a plain text format. To do this, choose "Text file" in the
output format listbox.
We have conducted a
large scale evaluation of the performance of the MHC class I binding predictions and found
that they in general rank as 1) ANN and 2) SMM method. Supplementary information, including
all datasets used to establish and evaluate the methods, is available
here.
By default, the overall best method (ANN) is selected. However, not all methods
can currently make predictions for all allele and peptide length combinations, so only the
methods available will be displayed.
As of April 2013, the tools have been trained on the new set of peptide binding data that
became recently available.
Thus, predictions made using the new tools may be different from those of the old tools.
• Sending the result table in a email:
Inputting your email address is recommended to ensure you could receive the result, especially for those prediction jobs which will take a long time. A email with the result table attached will send to you mail box as well as the result displayed on the web site.
One or multiple email addresses with comma separated could be accepted.
Example:
youremail@example.com
email1@example.com, email2@example.com, email3@example.com ...
Please input your email address for the extremely large predictions because for these jobs we only send the result to users by email. Or download the
standalone to finish these predictions locally.
For additional information regarding how to input your email address in the mhci API, Please look at the help page
here.
6. Submit the prediction
This one is easy. Click the submit button, and a result screen similar to the one below should appear.
Interpreting prediction output
Below is a screenshot of a prediction output page, with four relevant sections
marked that are described in more detail below.
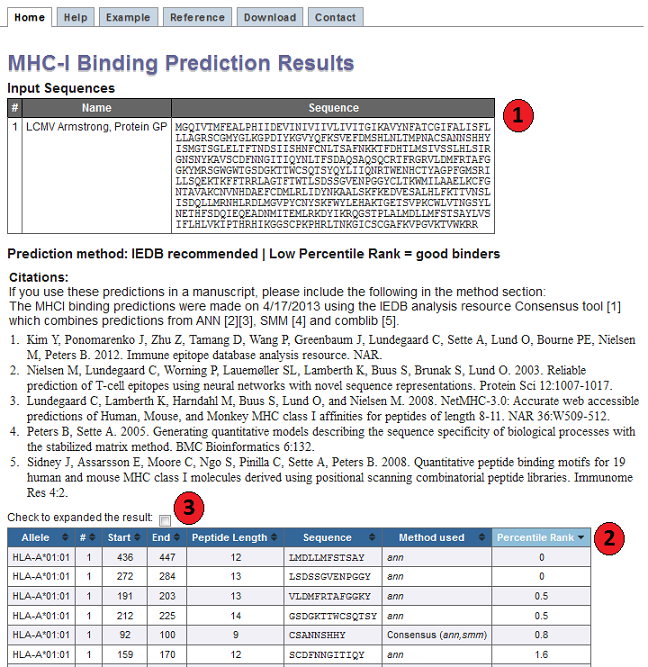
1. Input Sequences
This table displays the sequences and their names extracted from the user input.
If no names were assigned by the user (which is only possible in FASTA format),
the sequences are numbered in their input order (sequence 1, sequence 2, ...).
2. Prediction output table
Each row in this table corresponds to one peptide binding prediction. The columns contain the allele the
prediction was made for, the input sequence number (#), start position and end position of the peptide,
its length, the peptide sequence, ('method used' if IEDB recommended method is used and 'percentile rank' for both
IEDB recommended and consensus), the predicted affinity and precentile rank for ann, smm and comblib_sidney2008.
The table can be sorted by clicking on the table column headers.
3. Interpreting predicted affinities and percentile ranks
The predicted output is given in units of IC50nM. Therefore a lower
number indicates higher affinity. As a rough guideline, peptides with IC50
values <50 nM are considered high affinity, <500 nM intermediate affinity and <5000
nM low affinity. Most known epitopes have high or intermediate affinity. Some
epitopes have low affinity, but no known T-cell epitope has an IC50
value greater than 5000.
While the output of the predictions is quantitative, there are systematic deviations
from experimental IC50 values. For example, the makeup of the training
data and the prediction methods used have a non-trivial impact on the range of
predicted IC50 values. A detailed evaluation of the correlation
between predicted IC50 and antigenicity of peptides is currently being conducted
which will help to better interpret prediction results.
In addition to the IC50 values for each peptide, a percentile rank is generated by comparing the peptide's IC50
against those of a set of random peptides from SWISSPROT database. A small numbered percentile rank indicates high affinity.
For the 'consensus' and 'IEDB recommended' methods, the median percentile rank of the methods used is reported as the representative
percentile rank.
Regarding the peptides selection of the latest percentile data calculation, we downloaded the "Reviewed (Swiss-Prot)" dataset from https://www.uniprot.org/downloads in FASTA format on 10/29/2018. The file contained 558,712 sequences. 555,970 of them were acceptable by tools of IEDB, and 544,147 of them had length of at least 50 aa. Then we randomly selected 10,000 of these protein sequences and further randomly selected peptides with specified lengths (8-15 for class I; 10-30 for class II) from each protein sequence. Click
here to download the datasets.
4. Expanded result:
By default prediction result is collapsed to show only the percentile rank when either 'IEDB recommended' or 'consensus' method is used.
The table can be expended to display the individual score of different methods used by checking box above result table.
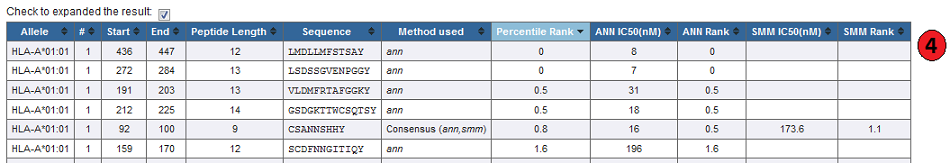
5. Default result:
Methods other than the IEDB recommended and Consensus, display just their IC50, or prediction scores.

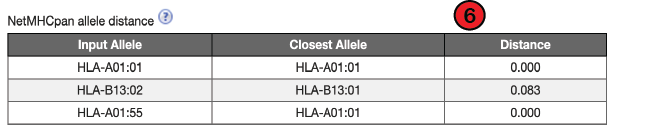
6. NetMHCpan allele distance:
The allele distance from the training data is the sequence distance of the predicted allele from its nearest neighbor in the training set. Alleles with lower distances to the training set will have more accurate predictions. A distance of 0 indicates a perfect match between alleles and values at or below 0.1 is considered acceptable for generating accurate predictions.